
How can DNA be damaged?
Did you know our DNA is continually being damaged throughout our lifetime?
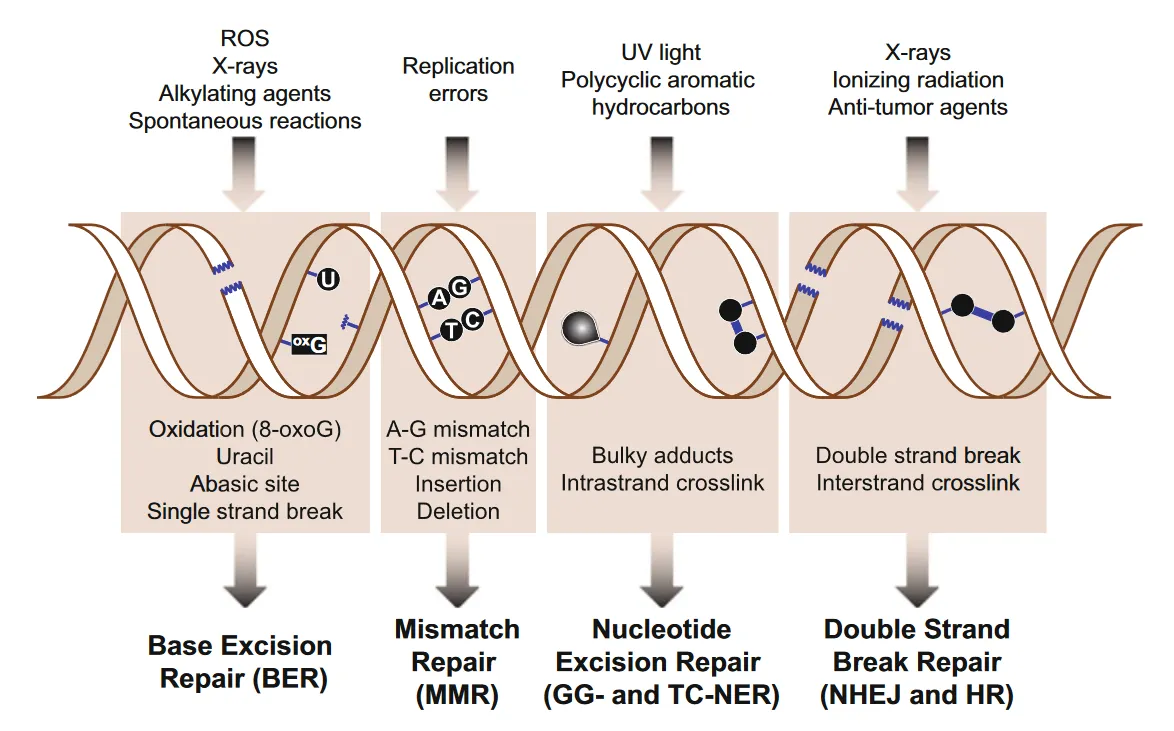
You are probably already aware that exposure to X-rays or nuclear radiation is capable of mutating our DNA, but normal cellular processes can also damage our DNA.
When cells replicate, for example, they can introduce mistakes into the DNA code, such as unnecessarily inserting or deleting nucleotide bases (the letters A, C, G, T) or by incorrectly pairing bases with one another (known as base mismatch).
As we’ve seen in several previous traits (such as your Oxidative Stress (SOD2) trait), cell respiration, the process by which cells generate energy, can also cause damage to DNA. When cells respire, they generate reactive oxygen species (ROS) – harmful substances that can oxidise and thereby damage nucleotide bases. ROS are also capable of breaking one of the strands of our DNA molecule. This is called a single-strand break.
Source: Dexheimer T. (2013) DNA Repair Pathways and Mechanisms. In: Mathews L., Cabarcas S., Hurt E. (eds) DNA Repair of Cancer Stem Cells. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4590-2_2
In addition to normal cellular processes within the body, substances and radiation in our external environment can also damage our DNA. For example, UV (ultraviolet) radiation in sunlight can cause abnormal links between nucleotide bases that are next to each other on the same DNA strand. This is known as an intrastrand crosslink. Typically, UV light can create an abnormal crosslink between two thymine (T) bases (forming what is known as a pyrimidine dimer).
Ultraviolet radiation can also cause breaks to both DNA strands in what is known as a double strand break. Other forms of ionising radiation (e.g. X-rays) can also cause double strand breaks.
KEY POINTS
- DNA can be damaged by both normal cellular processes (e.g. cell replication, respiration) and environmental factors (e.g. exposure to ionising radiation).
- There are different types of damage the DNA molecule can sustain e.g. single-strand breaks, double-strand breaks, base insertions, deletions, mismatch etc.
How is damaged DNA repaired?
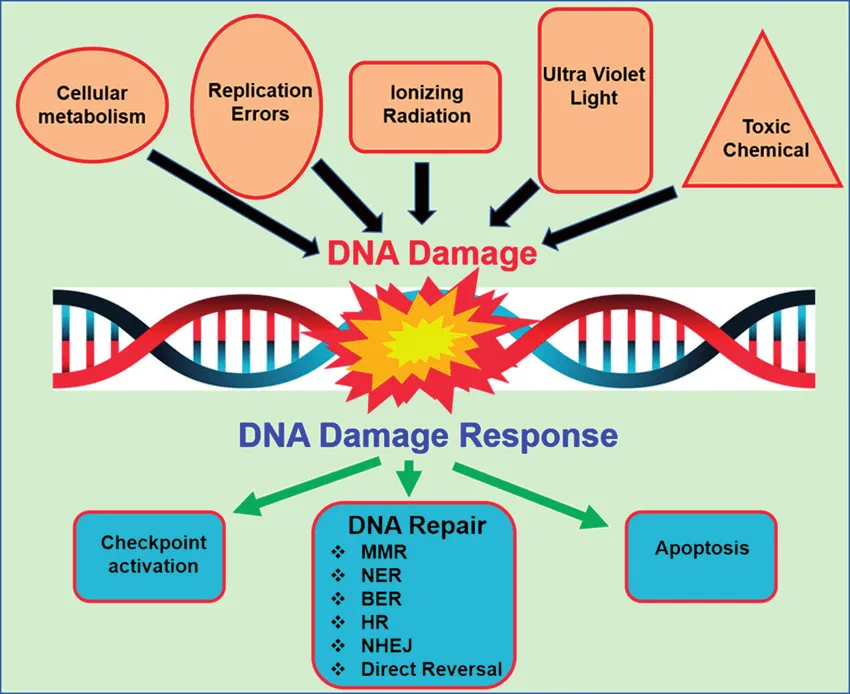
It’s thought that each cell in our body suffers between 1,000 and 1,000,000 lesions to DNA per day from both normal cellular processes and environmental factors. Given that DNA codes for and regulates proteins involved in lots of essential processes, the accumulation of DNA damage would, left unrepaired, impair normal cell function. Furthermore, as cells divide and replicate, such mistakes in the DNA code would be forwarded on to new cells.
Fortunately, we’ve evolved a host of repair mechanisms that can detect and fix damaged DNA. These are collectively known as the DNA damage response (DDR).
Source: Panneerselvam, J., Qu, D., Houchen, C., Bronze, M., & Chandrakesan, P. (2020). DCLK1 and DNA Damage Response. In Genotoxicity and Mutagenicity-Mechanisms and Test Methods. IntechOpen.
The exact type of DNA repair mechanism that is activated depends on the type of damage sustained. (They ways by which different enzymes removed, repair, and patch damaged regions of DNA are ingenious, albeit very complicated. Therefore, we’ll try to keep things simple and only cover things superficially).
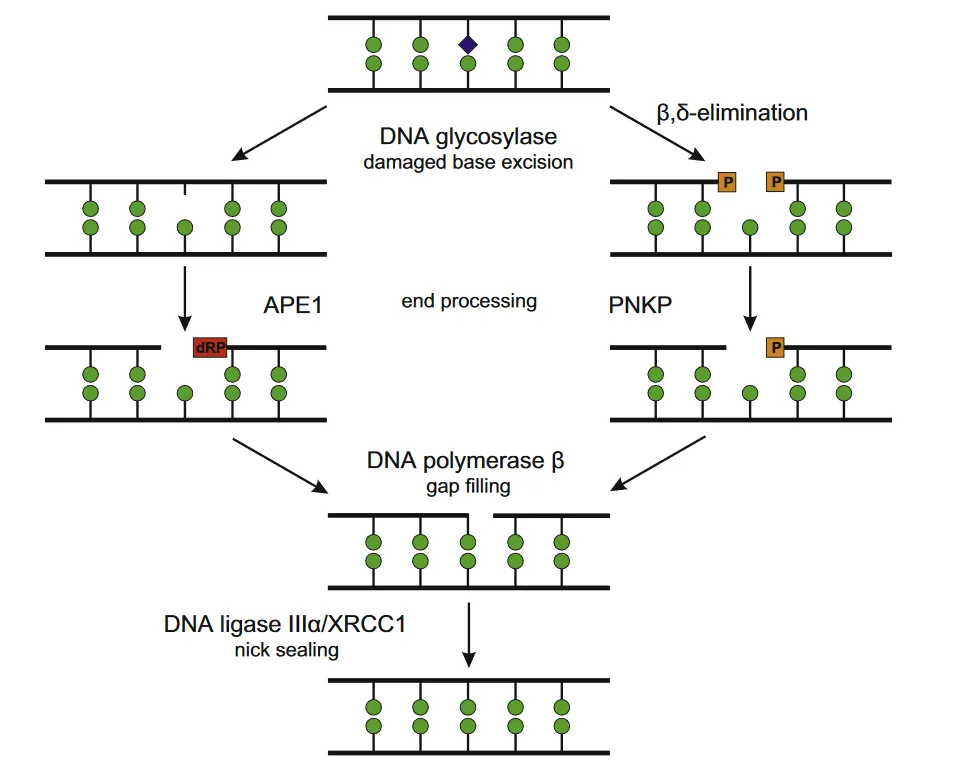
- Base excision repair (BER)
This kind of repair is used when small regions of DNA are damaged, but the overall shape of the DNA double helix molecule is not significantly distorted. For example, base excision repair may be used to repair single nucleotide bases that are damaged by reactive oxygen species (ROS).
As the name suggests, this repair mechanism involves removing or ‘excising’ the damaged base. Specialised enzymes make a cut either side of the damaged base on a DNA strand. The damaged base is then removed, the gap patched with new DNA, before the DNA strand is resealed.
Source: Edmonds, M. J., & Parsons, J. L. (2014). Regulation of base excision repair proteins by ubiquitylation. Experimental cell research, 329(1), 132-138.
Enzymes involved resealing the DNA strand in base excision repair are also used to repair single-strand breaks.
- Mismatch repair (MMR)
Mismatch repair (MMR) is used to correct mistakes introduced to the DNA sequence when cells replicate. As discussed in the previous section, during cell replication nucleotide bases may be erroneously inserted, deleted, or paired incorrectly with one another.
During mismatch repair, enzymes recognise the mismatched base pairs and proceed to remove the incorrect bases and their neighbours. They then patch the gap with the correct bases.
- Nucleotide excision repair (NER)
Nucleotide excision repair (NER) is used to fix damage such as intrastrand crosslinks caused by UV radiation. It involves recognising the abnormally crosslinked bases, removing them, and then filling in the gap with newly synthesised DNA.
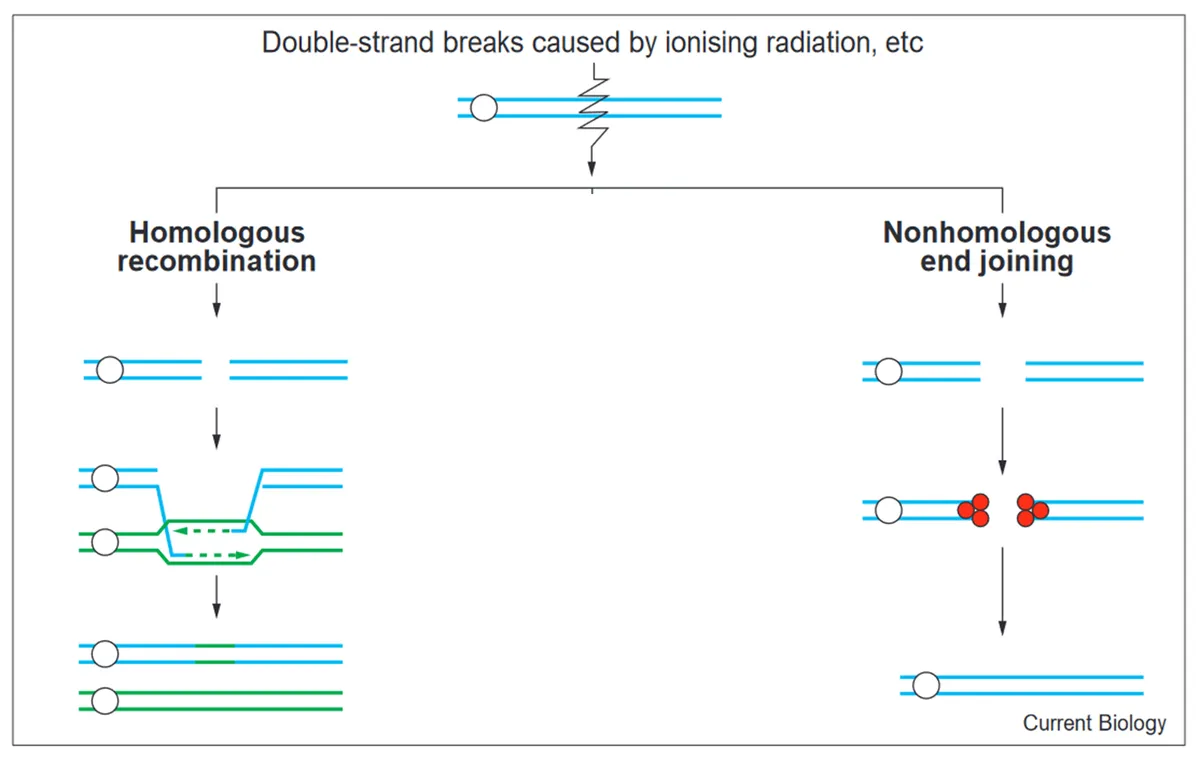
- Double strand break repair
Double strand breaks (DSBs), which involve breakages to both strands of the DNA molecule, have the potential to cause cell death if left unrepaired. Fortunately, we have evolved two main ways of fixing double-stranded breaks: non-homologous end-joining (NHEJ) and homologous recombination (HR).
NHEJ can be likened to gluing two broken ends together. Imagine if we broke a rod. To repair the rod, we could apply glue to both broken ends and then force them together again. This is essentially what happens in NHEJ - a protein called Ku is attached to both of the broken ends of the DNA molecule, and then the ends are reattached to one another. In the same way we may sand or file the broken rod ends to ensure a snug fit before reattaching, the broken ends of the DNA strand may be “cleaned” before reattachment. This may involve, for example, removing unnecessary bases, so that the two broken ends can be properly aligned.
When double-strand breaks occur, some nucleotides near the breakage sites may be lost. Using the broken rod analogy, this is like losing some shards or shavings at the broken ends. If you were to then glue both broken ends together in a roughr and ready fashion, you may be missing some bits of the original rod. Similarly, during NHEJ, nucleotide bases around the breakage site may be lost.
Source: Featherstone, C., & Jackson, S. P. (1999). DNA double-strand break repair. Current Biology, 9(20), R759-R761.
Homologous recombination (HR) is it a bit more complicated than NHEJ. To continue the broken rod analogy, it involves sourcing an identical rod and using this as a template to 3D print a joint between the two broken ends of the original rod.
In DNA terms, HR entails repairing a double strand break by inserting (or recombining) the broken DNA strands into an identical (homologous) piece of DNA (such as sister chromosome). The identical piece of DNA is then used to as a template to create a bridge between the ends of the originally broken DNA molecule.
As HR involves replacing the breakage with an identical piece of DNA, it does not lead to a loss of genetic information (unlike NHEJ).
Checkpoint activation and apoptosis
For us to grow and develop, and for our organs and tissues to function effectively, their constituent cells need grow and divide. Cells go through a series of phases, known as the cell cycle, whereby they enlarge in size, replicate their DNA, and then ultimately divide into two new cells. They then begin a new cycle.
The cell cycle has a number of checkpoints whereby it “decides” whether it should progress to the next phase and start dividing. These checkpoints are particularly important in the context of DNA damage. If a cell with considerable DNA damage is allowed to pass through unchecked and divide, it may pass on harmful DNA mutations to future generations of cells that, more widely, disrupt tissue and organ function.
Therefore, in response to DNA damage, cells may be ‘arrested’ at a particular checkpoint and not be allowed to divide and replicate. The cell may also be eliminated by entering a process of programmed cell death known as apoptosis.
KEY POINTS
- Cells activate a DNA damage response (DDR) when they sense damage to DNA molecules.
- The DDR includes various repair mechanisms to fix different types of DNA damage.
- Generally speaking, these repair mechanisms involve enzymes that remove damaged DNA bases, patch gaps with new, correct DNA, and then reseal any breakages.
- Cells may also be prevented from dividing and targetted for programmed cell death (apoptosis) in response to DNA damage.
What is the ATM gene?
The ATM (ataxia-telangiectasia mutated) gene encodes a protein that senses DNA damage and initiates repair mechanisms. It plays a particularly important role in the identification and repair of double-strand breaks to DNA.
The ATM gene is so-called because rare mutations within this gene can lead to a hereditary condition known as ataxia telangiectasia. This condition is characterised by progressive difficulty with movement and coordination (ataxia) and also a higher risk of cancers. The increased cancer risk in ataxia telangiectasia is likely due to impairments in DNA repair mechanisms, which allow other mutations to accumulate and cause uncontrolled cell growth.
These impairments in DNA repair result from rare mutations in the ATM gene that give rise to a non-functional ATM protein.
Please note that your ATM and DNA damage trait does not look at these rare mutations associated with a non-functional ATM protein and development of ataxia-telangiectasia. Instead, we look at more common ATM mutations (SNPs) that are linked to small differences in susceptibility to DNA damage.
KEY POINTS
- The ATM gene plays a important role in sensing DNA damage and activating DNA repair mechanisms.
- The ATM gene is particularly important for the repair of double-strand breaks.
- ATM gene variants can affect susceptibility to DNA damage.
How does ATM assist with DNA repair?
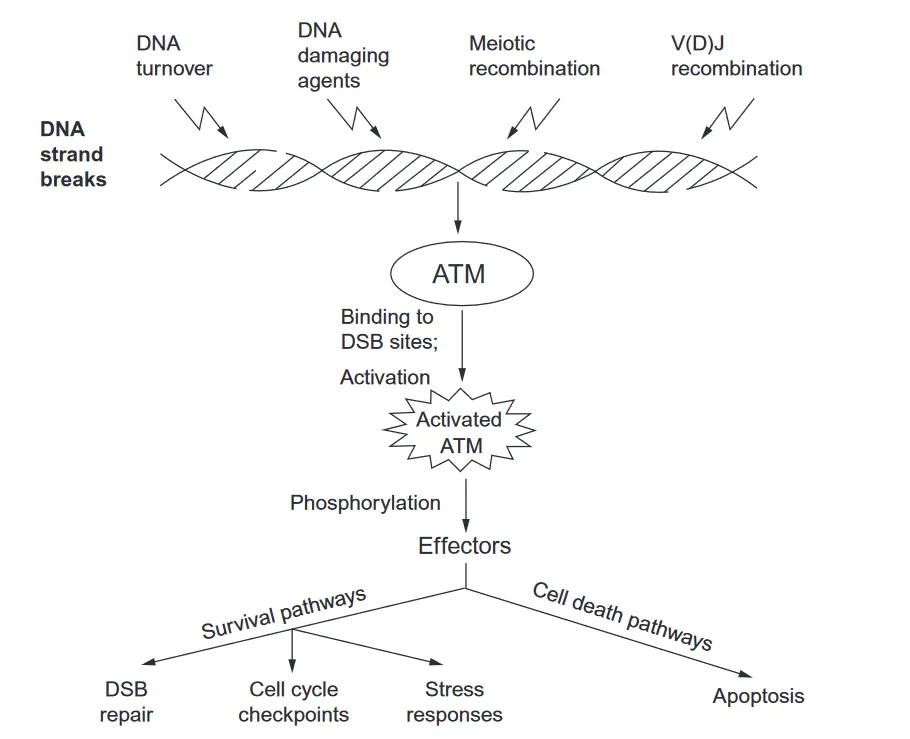
The ATM protein (which is encoded by the ATM gene) is an enzyme called a protein kinase. In response to DNA damage, this enzyme activates various substrate molecules (effectors) that carry out DNA repair and prevent damaged cells from replicating.
In particular, the ATM protein is recruited to sites of double-strand breaks (DSBs) in the DNA molecule. After binding to a breakage site, the ATM protein becomes activated and then stimulates other effector molecules that go on to repair the double-strand break.
For example, ATM can activate the enzymes involved in homologous recombination (HR) and non-homologous end-joining (NHEJ) of double-strand breaks (discussed in the previous section).
Source: Nissenkorn, A., & Ben-Zeev, B. (2015). Ataxia telangiectasia. Handbook of clinical neurology, 132, 199-214.
ATM also activates checkpoints to prevent cells with damaged DNA from replicating and dividing. In this respect, one of the key proteins activated by ATM is p53 – known as the ‘guardian of the genome.’ P53 is an example of a tumour suppressor protein – it stops cells with damaged DNA from progressing through the cell cycle and dividing. Without the action of ATM and p53, damaged cells may continue to replicate, leading to uncontrolled cell growth.
In addition to arresting damaged cells at checkpoints to prevent them from dividing uncontrollably, ATM can also target damaged cells to “commit cell suicide” through apoptosis – programmed cell death.
KEY POINTS
- The ATM protein binds to damaged DNA and activates other molecules responsible for DNA repair.
- ATM also activates cell cycle checkpoints to prevent damaged cells from dividing.
- ATM can also cause damaged cells to be eliminated by a process of programmed cell death (apoptosis).
How do ATM gene variants affect susceptibility to DNA damage?
There are several different SNPs (Single Nucleotide Polymorphisms) within the ATM gene that are posited to affect susceptibility to DNA damage and uncontrolled cell growth.
One of these SNPs, designated rs664143, causes an A à G change in the ATM gene, giving rise to two different ATM gene variants or ‘alleles’: the ‘A’ allele and the ‘G’ allele.
Of these, the ‘A’ allele has been associated with an increased susceptibility to DNA damage and uncontrolled cell growth.
Uncontrolled cell growth in lung tissue can give rise to lung cancer, and some meta-analyses have linked the ‘A’ allele to an increased risk of lung cancer. Studies in these meta-analyses tend to compare the ATM genotype of ‘cases’ with lung cancer to healthy controls. They then observe whether one particular genotype or allele is overrepresented in the lung cancer cases. More specifically they calculate an odds ratio, which grossly calculates how much more likely one genotype is to have lung cancer versus another genotype.
For example, a 2017 meta-analysis found that the AG and AA genotypes were associated with a odds ratio of 1.48 for lung cancer. This means that those with the AG and AA genotypes (i.e. ‘A’ allele carriers) were 1.48 times more likely (or 48% more likely) to have lung cancer compared to those with the GG genotype. This is illustrated in the Forest plot below.
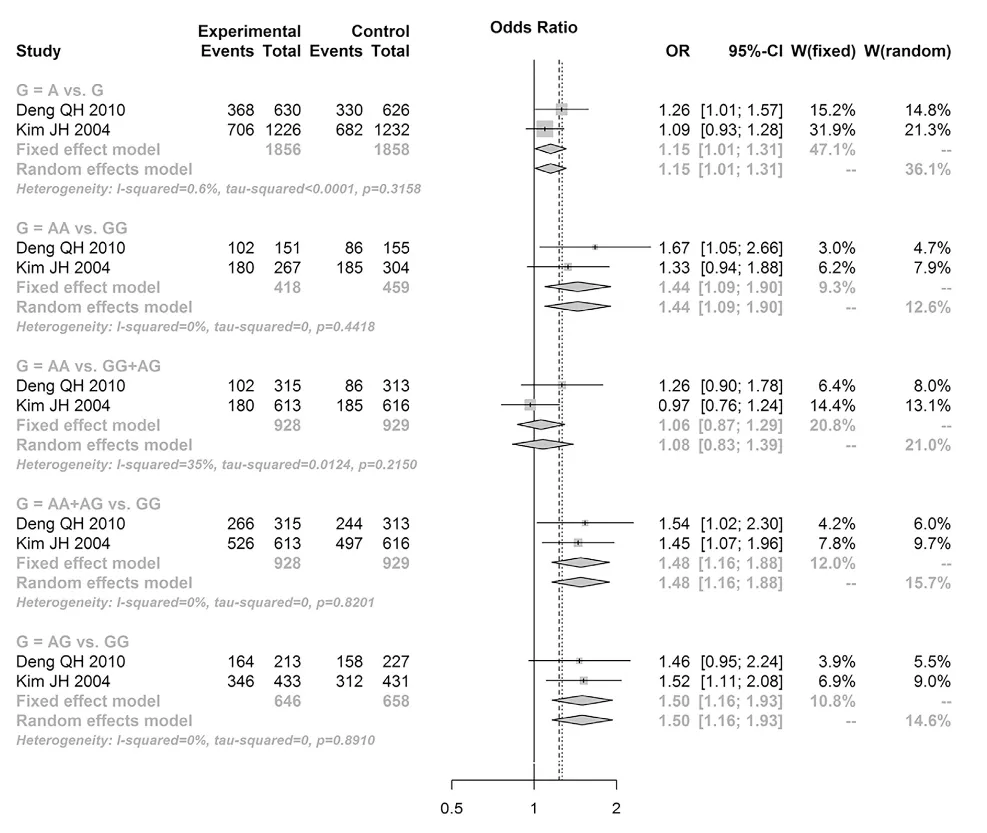
Source: Xu, Y., Gao, P., Lv, X., Zhang, L., Li, W., & Zhang, J. (2017). A meta-analysis of the relationship between ataxia-telangiectasia mutated gene polymorphisms and lung cancer susceptibility. Pathology-Research and Practice, 213(9), 1152-1159.
Note, however, that these odds ratios are associated with genotypes only and cannot be used to calculate individual risk of lung cancer. Furthermore, only very large odds ratios are useful as a marker of disease. For example, if you want to detect 80% of lung cancer cases based on the presence of the risk ‘A’ allele, then, with a low odds ratio of 1.5, about 73% of healthy controls would be incorrectly labelled as lung cancer cases.
It is also important to frame these genetic association studies in the context of other genes and environmental factors that affect lung cancer risk. Individual genes / alleles typically only explain a tiny proportion of variance in many diseases, including lung cancer. By contrast, smoking is estimated to explain 90% of variance of lung cancer risk in men. If you currently smoke, stopping smoking will arguably have the largest impact on your risk lung cancer.
The effect of antioxidant intake on DNA damage susceptibility
Some limited evidence suggests that higher intakes of antioxidant vitamins, including Vitamin A, Vitamin C, and folic acid, can moderate the effect of the ‘A’ allele on risk of DNA damage and uncontrolled cell growth.
In an analysis of subjects in the Korean Breast Cancer Study, researchers found that the ‘A’ allele (rs664143), as part of another set of coinherited SNPs in the ATM gene, was linked to a higher risk of breast cancer. This association was reduced in women with high intakes of Vitamin A, Vitamin C, and folic acid.
Further studies are needed in this area, but it is worth pointing out that a healthy diet (with a variety of plant-based micronutrients) and lifestyle are widely shown to help protect against the development of cancer, regardless of genotype.
In 2014, it was estimated that a 13.9% and 22.4% of cancers in US men and women, respectively, could be attributed to a combination of poor diet, excess body weight, physical activity, and alcohol consumption. In this respect, a diet rich in fruit and vegetables, a healthy body weight, regular physical activity, and moderated alcohol intake can all help to reduce cancer risk.
KEY POINTS
- The 'A' allele (rs664143) of the ATM gene has been linked to greater susceptibility to DNA damage and uncontrolled cell growth.
- Some studies have associated the 'A' allele with an increased risk of lung cancer but these studies must be interpreted with caution.
- Some limited evidence suggests higher intakes of antioxidant vitamins may help reduce cancer risk in 'A' allele carriers.
- Poor diet, physical inactivity, excess body weight and alcohol consumption can all increase cancer risk.
Your ATM and DNA damage trait
Your ATM and DNA damage trait looks at the rs664143 SNP in the ATM gene to analyse your susceptibility to DNA damage. Depending on your DNA results, you will be classified into one of the following two groups:
- Average – you do not carry the risk ‘A’ allele associated with increased susceptibility to DNA damage and uncontrolled cell growth. Your genotype is GG.
- Increased risk – you carry the risk ‘A’ allele associated with greater susceptibility to DNA damage and uncontrolled cell growth. You genotype is either AG or AA.
To find out your result, please login to truefeed.